Marfanův syndrom
Mareike Müller je nezávislá spisovatelka na lékařském oddělení a asistentka lékaře pro neurochirurgii v Düsseldorfu. Vystudovala humánní medicínu v Magdeburgu a během svého pobytu v zahraničí na čtyřech různých kontinentech získala mnoho praktických lékařských zkušeností.
Více o odbornících na Veškerý obsah je kontrolován lékařskými novináři.Marfanův syndrom (MFS) je genetické onemocnění pojivové tkáně. Pacienti mají různé příznaky v různé míře: dlouhé prsty a úzké, dlouhé končetiny nebo poškození cév. Na Marfanův syndrom neexistuje lék. Pravidelné kontroly mohou zabránit komplikacím. Přečtěte si vše o Marfanově syndromu zde!
Kódy ICD pro toto onemocnění: Kódy ICD jsou mezinárodně uznávané kódy pro lékařské diagnózy. Lze je najít například v lékařských listech nebo na potvrzeních o pracovní neschopnosti. Q87
Marfanův syndrom: popis
Marfanův syndrom je genetické onemocnění, které se přenáší buď z rodičů na dítě, nebo se vyvíjí spontánně. Nemoc, která se vyvíjí spontánně, je také známá jako sporadické onemocnění. To se týká asi 25 až 30 procent pacientů s Marfanovým syndromem. Celkově je Marfanovým syndromem postižen jeden až pět z 10 000 lidí v populaci. Mezi pohlavími není žádný rozdíl.
Marfanův syndrom: Příznaky
Známky Marfanova syndromu jsou u jednotlivých pacientů velmi odlišné a různě výrazné. I ve stejné rodině se příznaky Marfanova syndromu mohou mezi nemocnými členy rodiny velmi lišit. Nemoc je ovlivněna různými orgánovými systémy. Nejběžnější jsou změny
- Kardiovaskulární systém
- kostra
- oko
Marfanův syndrom: Kardiovaskulární systém
Pacienti s Marfanovým syndromem mají zvýšené riziko náhlé smrti. Důvodem je často se vyskytující trhlina ve stěně hlavní tepny (aortální disekce). V důsledku vytvoření mezery uvnitř stěny aorty již krev není transportována do menších krevních cév, ale spíše prosakuje do mezer. Riziko disekce aorty je u pacientů s Marfanovým syndromem zvýšeno, protože jejich aorta, která má oslabené stěny, se postupně rozšiřuje (progresivní aortální dilatace).
Pacienti navíc často trpí poškozením srdeční chlopně, jako je aortální a mitrální regurgitace. Ty mohou vést k srdeční arytmii. Dále jim hrozí zánět srdce (endokarditida) a srdeční selhání.
Marfanův syndrom: Kostra
Kosterní změny jsou často prvním znakem Marfanova syndromu. Pacienti vynikají vysokou postavou a velmi úzkými, dlouhými končetinami. Spider prstoklad (arachnodaktylie) je známým příznakem. Prstovým pavoukům se tak říká, protože jsou extrémně dlouhé a úzké.
Mnoho pacientů má navíc deformity hrudníku, jako jsou kuřecí nebo trychtýřová prsa. Jako další změny skeletu často trpí skoliózou, ohnutím a zkroucením páteře. Někteří pacienti mají navíc nedostatečně vyvinuté obličejové kosti, například lícní kosti nebo horní čelist.
Celistvost těchto kosterních změn je také známá jako marfanoidní habitus.
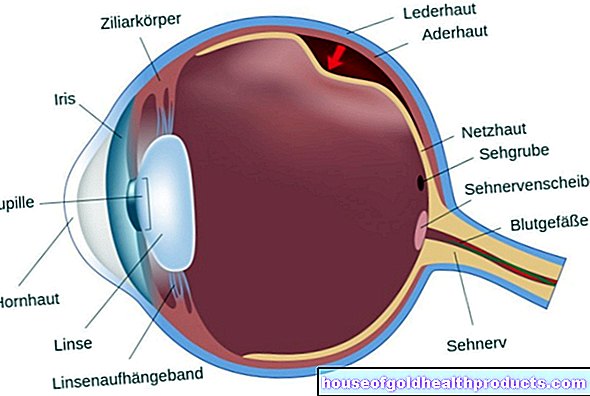
Marfanův syndrom: oko
Změny v oku způsobené Marfanovým syndromem postihují hlavně čočku. Často je posunutý (ektopie objektivu). To ohrožuje pacienta oslepnutím. Dalším rizikovým faktorem oslepnutí je krátkozrakost. Je to způsobeno příliš dlouhou oční bulvou. Tato změna může také vést k odchlípení sítnice.
Marfanův syndrom: příznaky postihující jiné orgány
Kromě zmíněných orgánových systémů může Marfanův syndrom poškodit i další struktury. Patří sem mimo jiné plíce. U postižených je zvýšené riziko vzniku pneumotoraxu. Lékaři to chápou tak, že to znamená odloučení plicní pohrudnice od pohrudnice a pronikání vzduchu do této mezery. Tento stav může být život ohrožující, protože v postižené oblasti se zhroutí plíce.
Strie jsou často pozorovány na kůži pacientů s Marfanovým syndromem jako znak slabé pojivové tkáně.
V průběhu života se může vyvinout takzvaná duraektázie. Jedná se o rozšíření mozkových obalů, obvykle na úrovni bederní páteře. Často je asymptomatický. V některých případech může způsobit bolest, když durální vak tlačí na vystupující spinální nervy.
Marfanův syndrom: příčiny a rizikové faktory
Marfanův syndrom je autozomálně dominantní dědičné onemocnění. To znamená, že v našem genetickém složení dochází ke změně (mutaci) genu, která spouští nemoc. Autosomálně dominantní popisuje, že tato genetická informace je umístěna na genově nespecifickém genovém komplexu (autozomálně) a vždy se objeví (dominantní).
Pokud má pacient s Marfanovým syndromem dítě, může zdědit buď nemocný, nebo zdravý gen. Protože každý člověk má dvojí sadu genetické výbavy. To znamená, že pravděpodobnost přenosu je 50 procent. Dítě pacienta s Marfanovým syndromem má 50 procentní pravděpodobnost onemocnění.
Marfanův syndrom: poškozená pojivová tkáň
Mutace, která způsobuje Marfanův syndrom, je na dlouhém rameni chromozomu 15 (15q21). Ovlivňuje takzvaný gen FBN1. Tento gen je zodpovědný za tvorbu proteinu pojivové tkáně, fibrillin-1. Fibrillin-1 je důležitý pro stabilitu pojivové tkáně. Pokud je jeho tvorba omezena mutací, pojivová tkáň ztrácí stabilitu.
Marfanův syndrom: různé formy
Závažnost Marfanova syndromu se liší. Lékaři pak hovoří o proměnlivé expresivitě. To znamená, že symptomy pacientů se v rámci rodiny také liší. Přes stejnou mutaci může mít pacient jen stěží nějaké příznaky, zatímco sourozenec ukazuje úplný obraz Marfanova syndromu.
Marfanův syndrom: Vyšetřování a diagnostika
Diagnózu Marfanova syndromu často stanoví dětský lékař. Celkově různí specialisté hrají roli v diagnostice, léčbě a poradenství. Kromě pediatra sem patří lidští genetici, kardiologové, ortopedové a oční lékaři. Před konečnou diagnózou se vás lékař nejprve podrobně zeptá na vaši anamnézu (anamnézu). Položí vám mimo jiné následující možné otázky:
- Má rodinný příslušník Marfanův syndrom?
- Cítíte občas závodní srdce?
- Byl jste v dětství vždy vyšší než ostatní?
- Jste krátkozrakí?
Fyzikální vyšetření Marfanova syndromu
Váš lékař poté provede fyzické vyšetření. Přitom se nejprve na kostru zblízka podívá. Věnuje pozornost délce jednotlivých kostí, tvaru hrudníku a tvaru obličeje. Poté poslouchá srdce a plíce. Nad hlavní tepnou lze zaznamenat srdeční arytmie nebo zvuky proudění.
Aby bylo možné diagnostikovat Marfanův syndrom, byla vyvinuta takzvaná Gentova kritéria. Uvádí různé příznaky onemocnění v různých formách. Když je splněn určitý počet kritérií, může být stanovena diagnóza.
Možný je také test genetického Marfanova syndromu. Analyzuje se genetická výbava a hledá se mutace, která je za nemoc zodpovědná. Pokud jsou v rodině případy Marfanova syndromu, lze před porodem stanovit příslušnou diagnózu.
Marfanův syndrom: Podobné klinické obrázky
Od Marfanova syndromu je třeba odlišit další genetická onemocnění, která mohou vést k podobným symptomům. Mezi ně patří mimo jiné
- Ehlers-Danlosův syndrom
- Loeys-Dietzův syndrom
- Sphrintzen-Goldbergův syndrom
- MASS syndrom
Marfanův syndrom: Léčba
Vzhledem k tomu, že Marfanův syndrom je genetickým onemocněním, samotnou příčinu, mutaci, nelze léčit. Cílem terapie je pravidelná kontrola pacienta různými specialisty, aby se předešlo komplikacím. Nejdůležitější je monitorování srdce, aby se zabránilo náhlé smrti z aortální disekce. Toho lze dosáhnout působením proti aortální dilataci podáním beta blokátorů a omezením fyzické aktivity. Dilataci aorty lze sledovat každoročními ultrazvukovými vyšetřeními a kořen aorty lze včas opravit před pitvou.
Další operace, které mohou být nutné v případě Marfanova syndromu
- Korekce skoliózy
- Korekce hrudníku
- Vyjmutí objektivu
Marfanův syndrom: průběh nemoci a prognóza
Pravděpodobnost přenosu mutace z jednoho rodiče na dítě je 50 procent. Páry s partnerem s Marfanovým syndromem, které plánují mít děti, by měly požádat o radu lidského genetika.
Dnes je délka života a kvalita života u pacientů s Marfanovým syndromem téměř neomezená. I když byla délka života v minulosti jasně omezená, za posledních 30 let se prodloužila o 30 let. U pacientů je však stále zvýšené riziko aortální disekce, která může mít za následek náhlou smrt. Disekce aorty je nejčastěji pozorována kolem 30. roku života. Pravidelné kontroly ošetřujících specialistů mohou snížit riziko disekce aorty u Marfanova syndromu.
Tagy: zdravé nohy sportovní kondice zprávy